
获得全基因测序的最优方案—低深度重测序(LcWGS)有奇招!
全基因组关联研究(GWAS)已经确定了数千种与人类和农业物种复杂性状相关的遗传变异。尽管测序成本不断下降,大规模的全基因组测序(几千个样本)的成本仍然很高。在许多情况下,基于参考面板填充的策略将低密度数据填充为高密度数据,为全基因组关联分析提供了一种经济有效的方法。
在过去十年中,产生了越来越多样本的参考面板(such as 1000 Genomes (1000G),Haplotype Reference Consortium (HRC),and the TransOmics for Precision Medicine (TOPMed) Program)。参考面板样本量的连续增加促使我们捕获了更多罕见的变异,并在关联研究中提供了更高分辨率的图谱。但是随着面板的增加,如何选择最佳参考面板也是一个挑战。例如,当在特定研究群体中(如撒丁岛、芬兰、挪威、和冰岛等地进行的研究),较小的定制参考面板可能要比使用广泛使用的公共参考面板有更好的填充效果。但是定制的参考面板会可能会遗漏一些罕见的突变和单倍型,而这些突变和单倍型同样也可能会被较大的面板所覆盖,并且可能对具有独特祖先的个体表现不佳。理想的解决方案是构建一个组合参考面板。
2022年5月3日,密歇根大学生物统计学系Gonçalo Abecasis研究员与他的研究团队在AIHG发表"Meta-imputation: An efficient method to combine genotype data after imputation with multiple reference panels"文章中提出了一种Meta-imputation方法,该方法允许将使用不同参考面板生成的多个填充结果合并到一起,生成一致填充数据集。
Meta-imputation包括两个单独的步骤(minimac4+MetaMinimac2),即填充和整合,如下图:

meta-imputation分析流程图
首先,根据两个或多个不同的参考面板估算目标样本。文章中创新的地方是使用每个基因组中的基因型标记来估计每个个体的局部权重。然后通过依次掩蔽每个观察到的基因型,试图根据侧翼标记的信息对其进行填充,文章中称此程序的估算结果为leave-one-out (LOO) dosages。
通过比较LOO dosages和掩蔽位点的原始基因型来评估每个参考面板的局部填充性能,并相应地分配局部权重。如下图中展示了使用两个参考面板的LOO填充算法的简化版本。

leave-one-out填充示例图
为了评估Meta-imputation对于填充混合个体基因组的能力,文章中选择了一组1000G具有混合祖先的样本,并创建了两个填充面板——一个具有大部分欧洲血统的个体(EUR,503名欧洲人),另一个具有大部分非洲血统的个体(AFR,600名非洲人)。将美国西南部(ASW)的61名非裔美国人(采用Illumina Human1M-Duo BeadChip (19,883 out of 1,803,869 variants on chromosome 20)获取基因型),使用欧洲面板和非洲面板进行了Meta-imputation,并通过计算 imputed results和masked genotype data之间的aggregated r2来评估填充准确性,如下图:

非裔美国人样本填充精度比较图
从结果来看,Meta-imputation的准确度大大高于使用单一参考面板进行填充的准确度。对于MAF为0.05%~0.1%,与单独使用AFR面板(r=0.313)或单独使用EUR面板(r=0.009)的填充相比,Meta-imputation实现了更高的精确度(填充剂量和实际基因型之间的r=0.427),并且Meta-imputation的精确度与使用AFR+EUR面板(r=0.425)的精确度相当。总的来说,与使用一个较小的面板相比,对于罕见变异,Meta-imputation具有最大的优势。
文章中还使用大小差异很大的参考面板,在南亚血统样本中评估Meta-imputation方法,如下图,采用TOPMed release 2 面板(包括97256个个体)和1000G phase 3(GRCh38)面板(包括2504个个体)对762个南亚样本进行填充。采用UK Biobank发布了约50000名个体的完整外显子组测序数据集作为真值集,以评估填充的准确性。从结果来看,Meta-imputation的准确度同样高于2个参考面板单独进行填充的准确度。

南亚样本填充精度比较图
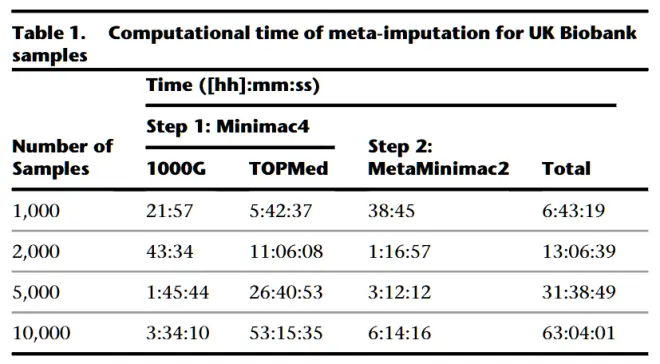
文章中还展示了Meta-imputation填充性能的优势,如下表,分别统计了1000、2000、5000和10000个目标样本的Meta-imputation单核计算时间。
结 论
Meta-imputation程序包括两个单独的步骤,即填充和整合,允许研究人员逐步考虑新的参考面板,而无需使用之前的面板重复填充步骤。随着每个面板的添加,研究人员只需针对新面板填充目标样本,然后即可将结果与之前计算的填充结果数据集相结合,实现面板共享,扩大了对越来越罕见变异的填充范围。除此之外,作者也指出了该方法的一些局限性,例如Meta-imputation是基于每个单倍型进行的,因此其性能取决于pre-phasing的质量。phasing中的switch errors可能导致填充精度降低和权重误导,因此Meta-imputation应直接受益于不断发展的phasing算法。
原创声明:本文由欧易生物旗下子公司青岛欧易报道,本文著作权归文章作者所有。欢迎个人转发及分享,未经作者的允许禁止转载。
