
每一次困惑,是一次升华的契机
激励我不断通过自媒体做视频教学,无论是短视频,还是小班课,就是有同学持续不断地深入学习,并一直保持专业的交流,在他们的研究中也派上了用场。今天我分享一个表面吸附小班的同学小叶带来的案例,我相信对于从事表面化学键分析的朋友们,非常有帮助。
首先看问题:H2分子键合非常简单,H-H之间有一个σ单键,成占据态(填入两个电子),而紧随它但能量更高的就是其反键态σ*。看起来非常简单的事情,但问题就在于小叶他做出来是这个样子(-COHP计算,右侧代表成键,左侧则是反键):
可以清楚看到,他得到的成键态是部分占据。这明显不复合H2的轨道特征。但错在哪里呢?如此简单的体系,计算参数都很规范,为甚么会得到这样的结果?为了搞清楚这一点,我们需要介绍一个概念 – 离散能级连续化。因为态密度是电子状态在能量空间的分布,但出现相邻状态都是高度局域的,而且彼此远离(即能级差很大),在数学上做连续化时容易显著偏离原来离散能级中心。我为了把这个例子凸显明显,在态密度小班课中特意设计了一个单Na原子态密度计算,如下图:

可以清楚看到,最内层轨道对应的能级计算值在-1031.78 eV(VASP,PBE泛函计算), 而实际做出来的态密度图在-1029.70 eV, 偏差幅度达到2 eV, 峰宽化也很明显。这是明显错误的,原因就在于它的下一个能级在1000 eV外,并且拥有更多的电子状态,因此连续化函数拟合时为了迁就高能级而移动了这个低能级以降低拟合偏差。但是这种处理毫无价值,因为我们关心的是费米能级附近的状态,对于深能级并不关心。
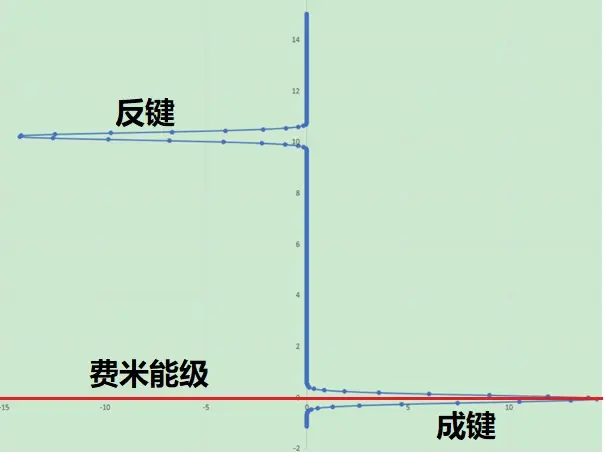
解决办法:当我们需要获得高品质态密度数据,需要密切注意你需要分析的电子状态大约在什么能量范围(一般都在费米能级附近),在VASP中就可以通过设置EMIN, EMAX, NEDOS来限定这个连续化时品质和能量分辨率(CASTEP中可以通过设置DOS范围来实现调控)。下面是小叶设置了以上参数新得到的H2分子的态密度图(COHP,没有加负号,所以左侧是成键、右侧反键):

必须恭喜小叶顺利得到可靠结果,也为他的认真点赞。各位朋友可能感受到,做理论计算分析需要非常细心,同时需要掌握的知识技能很多,马虎不得。
