
Gaussian快速启动教程(1):优化结构和计算单点能
前言
Gaussian软件有Windows版和Linux版,两版在使用上差别不大,其中虚拟机Linux版安装教程如前文所示(https://www.bilibili.com/read/cv17234114)。从本帖开始,记录和分享Gaussian软件的使用。所谓“快速启动教程”,即以计算目的为导向,快速上手软件。至于各种参数的意义和高级设置,则可以边用、边查、边学。
1. 基础概念
(1) 何谓“单点能”?
分子的一种构型构象,就是这个分子的一个“点”,该构型构象对应的能量就是它的“单点能”。分子的单点能随构型构象发生变化,据此可以绘制“势能面”。换句话说,“单点能”的“点”就是“势能面”上的一个点。
(2) 结构优化是做什么的?
结构优化,就是给定初始结构后,通过迭代计算,使分子优化至一个低单点能的“点”(即构型构象)。计算的初始参数要根据目标分子和计算机情况设置。迭代的收敛判据是对比上一次迭代和下一次迭代的计算结果,若能量(或分子内受力)小于某个值,则认为收敛。收敛结果有可能是整个势能面上的最低点,也有可能只是一个相对的最低点,因此初始结构的给定十分重要。
2. 绘制初始结构
初始结构可以通过GaussView面板手动绘制,也可以从外部导入。前者适合画简单分子,后者适合画复杂结构。
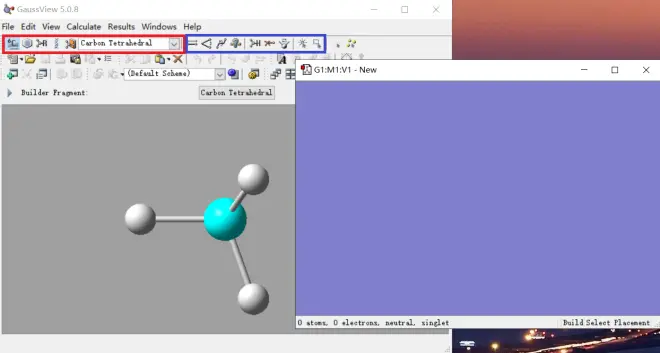
(1) 通过GaussView面板绘制
如图1,先通过主面板红框内的按钮选择要绘制的片段,然后在蓝色面板上绘制。若要改变已绘制结构的键长和键角等,可通过主面板蓝框内的按钮修改。每个按钮的具体功能不再赘述,挨个尝试几次即可熟悉。
(2) 通过外部导入(.mol格式文件)
GaussView自带面板有时不太直观,绘制复杂分子时容易出错。而且若对绘制面板不熟悉,绘制出来的初始结构有可能扭曲太大,不利于后续优化。因此通过第三方软件或网站绘制,再导入Gaussian是个不错的选择。个人推荐如下:
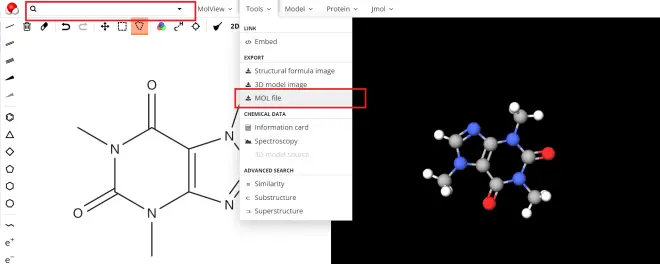
MolView网站(https://molview.org/):如图2,在左侧绘制点线式结构式后,右侧自动给出三维构型,通过上方按钮可以导出.mol格式文件。若分子是常见分子,可以直接在网页左上方搜索分子名。优点是十分直观,简单易用;缺点是网站服务器位于国外,有时打开可能较慢。
InDraw网站(http://indrawforweb.integle.com/):可以绘制点线式结构,然后导出.mol格式文件。优点是作为国内网站响应较快;缺点是需要注册登录使用,且没有直观的三维输出结果,没有分子数据库,遇到手性基团时也要小心绘制。
Materials Studio软件:这是一款强大的计算化学软件,收费软件,具体使用不再赘述,其“Clean”功能有类似优化的效果(Gaussian里也有,在右击“Edit”菜单里)。
T.E.S.T.软件(https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test):美国环保署(EPA)开发的基于构效关系(QSAR)的毒性预测软件,可免费使用。右侧可以绘制点线式结构,左上角也可以直接搜索分子名称,最后导出.mol格式文件。
其他类似网站和软件还有很多,不再赘述。
3. 启动软件
启动GaussView后打开初始分子结构的文件,另存为.gjf格式,注意路径不能有中文(不另存也没关系,后续开始计算也会提示保存)。
(1) 提交计算任务
在分子结构面板上右击 – Calculate – Gaussian Calculate Setup – 新窗口设置参数(如下文) – Submit(提交计算任务) – Save(保存初始文件) – 选择合适的目录和命名 – Save – 新窗口点击Yes。
Job Type – 任务类型。优化分子结构选Optimization或Opt+Freq(优化结构并计算虚频,之所以计算虚频,是因为若虚频不为零,则并非稳态分子);计算单点能选Energy。
Method – 第一行:Method第一项为目标分子的态,一般默认选Ground State(基态);第二项为计算方法,近年常用DFT(密度泛函理论);第三项为自旋,若不懂如何设置则先默认不改,计算出问题时再选择高线态(N线态的计算方法是N=2n+1,其中n是分子的孤电子数);第四项为所用泛函,近年常用B3LYP(计算方法和泛函的选项越靠下越精确,但相应耗时越长)。第二行:Basis set为基组,根据原子序数选择,越重的原子对应选项越靠下(具体可查询Gaussian官方手册)。
Title – 设置计算任务的名称。
Link0 – 设置计算任务所占用的计算机资源(不设置就会溢出报错)。Options选项卡:Memory Limit为最大占用内存,根据虚拟机系统占用内存规划,要为虚拟机系统预留一定内存(Windows版Gaussian09最大只能设为1G);Shared Processors为计算线程,一般设为虚拟机占用CPU核数,虚拟机占用核数一般设为计算机CPU核数;Chkpoint File为检查点文件,自行设置其保存位置和名称。上述设置对应代码见于Link选项卡。
上述内容可以写为默认设置。以Linux版为例,创建“Default.Route”文件,用记事本打开,写入如下内容,保存至“/home/lc/lc/g16/”路径(这是我的安装路径,读者需自行修改)。其中P和M分别代表调用CPU核数和内存大小,根据硬件和虚拟机设置。Windows版则为“Default.Rou”文件,保存在“C:\G09W\Scratch”路径。
-P- 4
-M- 8GB
Solvent – 选择溶剂。
Preview – 预览输入文件内容(如图3)。其大致由三块组成:第一块为计算任务相关信息,第二块为初始原子坐标,第三块为初始原子键连方式。第三块内容有些人会删除,因为结构优化后会自动给出。

(2) 查看计算进程(可选操作)
Windows版开始计算后会自动弹出Gaussian面板,显示计算进程,且不能关闭。而Linux版看不到这个面板,若要查看计算进程,可通过如下方法:
点击GaussView面板上的Calculate – Current Jobs
在新面板上选中任务 – Stream Output File
(3) 查看计算结果
计算完成时会自动弹出计算结果,若要打开其他文件,则点击File – Open – 选中输出文件 – 勾选最下方按钮 – Open
查看结构优化结果:新窗口右击 – Results – Summary
查看结构优化过程:新窗口点击绿色圆点
查看单点能优化过程:新窗口右击 – Results – Optimization
(教程结束)
