
深度剖析:理论与计算化学领域期刊中的“扛把子”——JCTC
Journal of Chemical Theory and Computation(以下简写JCTC),作为专门为理论与计算化学领域单独开辟的期刊,近些年影响因子也是逐年增加(近些年有些微波动,但范围较小;即时影响因子为5.011),大有成为理论与计算化学领域期刊中的“扛把子”。
JCTC发表论文、报告,包括量子电子结构、分子动力学和统计力学方面的新理论、方法论和/或重要应用等。特定的主题包括量子力学从头算或应用程序的发展,密度泛函理论,设计和新材料的性质,表面科学、蒙特卡罗模拟,溶剂化模型,QM / MM计算,生物分子结构预测以及分子动力学在最广泛的意义上包括气相动力学,从头开始动力学,生物分子动力学和蛋白质折叠等。遗憾的是,该期刊现已不考虑接受直接应用DFT和分子动力学等已知方法的论文。该杂志倾向于提交包括理论或方法的进展,并应用于引人注目的问题。

今天,我们就一起来剖析一下,JCTC在2020年发文情况,以及理论计算化学在这一年内的发展情况(注:此文中对JCTC期刊的分析数据,来源于Elsevier旗下的Scopus数据库,有疏漏之处,欢迎指出!)。
发文量
2020年JCTC共接受了645篇文章,比2019年多了80篇,其中文章类型方面,“article”共644篇,“Review”只有1篇。
发文归属地
排在第一位的仍是“United States”,全年在期刊上共发表了296篇,遥遥领先于排在第二位的“Germany”(共96篇),第一名是第二名的3倍多。而作为科研大国,中国全年在该期刊仅发表了48篇,可见国人对基础科学研究的重视程度,远远不及美国和德国。(其他排名详见图1)

图1 2020年JCTC期刊发文总数归属地排名(前10名)
发文归属机构
排在第一位的是“CNRS”,即法国国家科学研究中心,全年共发表了30篇文章;排在第二位的是“Lawrence Berkeley National Laboratory”,但仅有17篇,值得注意的是,发文归属机构前十名中,来自美国的共5位。(其他排名详见图2)
图2 2020年JCTC期刊发文总数归属机构排名(前10名)
个人发文总数
排在第一位的是来自于美国明尼苏达大学双城分校的Truhlar, D.G.,全年共发文9篇,发文之高,令我等望尘莫及。排在第二位的,同样是来自于美国,加州大学伯克利分校的Head-Gordon, M. (共6篇)。同样令人诧异的是,个人发文总数前十名的,总共有7人来自于美国。(其他排名详见图3)
图3 2020年JCTC期刊个人发文总数排名(前10名)
关键词出现次数
去除一些与理论计算化学无关的关键词,出现次数最多的关键词是“Molecular Dynamics”(以下简称MD),即“分子动力学”,共出现57次。由此可见去年一年全世界的科学家,在此领域付出的心血和努力,同时也可看出,“MD”将是未来理论计算化学的重要发展方向。(其他排名详见图4)
图4 2020年JCTC期刊关键词出现次数排名(前10名)
由于发文数量众多,我们这里选择了被引次数排名前十的文章进行阐述,本期我们先来了解一下被引次数排名前五的文章。
1.ff19SB:根据溶液中的量子力学能面训练出的氨基酸特异性蛋白骨架参数
分子动力学(MD)模拟在研究生物分子的运动和功能方面越来越受欢迎。然而,模拟的准确性在很大程度上,取决于分子力学(MM)的力场(FF),这是一组具有可调参数的函数,可以从原子位置计算势能。然而,FF的整体质量,如之前发布的ff99SB和ff14SB,可能会受到多年前假设的限制。在此,来自美国布鲁克海文国家实验室的Qin Wu&美国石溪大学的Carlos Simmerling等研究者,提出了更新版本的模型(ff19SB),研究者显著改进了所有20种氨基酸的主干结构。研究者利用二维量子力学(QM)能量面作为参考数据,利用多种氨基酸的二维φ/ψ构象扫描,拟合了耦合的φ/ψ参数。通过在水溶液中使用QM/MM来解决二面体参数拟合过程中的极化不一致性。最后,研究者分析了骨架拟合侧链转子的可能依赖性。为了广泛验证ff19SB参数,并与使用其他Amber力场的结果进行比较,研究者在显式溶剂中进行了总计约5 ms的MD模拟。研究结果表明,在对QM数据进行溶剂极化的氨基酸特异性训练后,ff19SB不仅能更好地重现氨基酸特异性蛋白数据库(PDB) Ramachandran图的差异,而且在区分氨基酸依赖性质(如螺旋倾向)方面也有显著提高。研究者还得出结论,在ff14SB中存在螺旋度的固有低估,这(不确切地)可由TIP3P偏向于过紧结构驱动的螺旋含量的增加来补偿。综上所述,ff19SB与更精确的水模型如OPC结合,在建模序列特异性行为、蛋白质突变和合理的蛋白质设计方面应该有更好的预测能力。

参考文献:Chuan Tian, Koushik Kasavajhala, Kellon A. A. Belfon, Lauren Raguette, He Huang, Angela N. Migues, John Bickel, Yuzhang Wang, Jorge Pincay, Qin Wu, and Carlos Simmerling. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. Journal of Chemical Theory and Computation 2020 16 (1), 528-552
DOI: 10.1021/acs.jctc.9b00591
原文链接:
https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b0059
2.激发态的登山策略:中等大小分子的高度精确的能量和基准

此文中,来自意大利比萨大学的Filippo Lipparini和法国国家科学研究中心对的Pierre-François Loos&Denis Jacquemin等人,给出了27个分子的高度精确的垂直跃迁能,可能包括有4,5,和6个非氢原子:丙酮、丙烯醛、苯、丁二烯、氰乙炔、氰甲醛、氰原、环戊二烯、环丙烯、环丙烯乙酮、二乙炔、呋喃、乙二醛、咪唑、异丁烯、亚甲基环丙烯、丙烯、吡嗪、吡啶、嘧啶、吡咯、四嗪、硫代丙酮、噻吩、硫代丙烯、三嗪。为了获得这些能量,对于这些系统,研究者使用运动方程/线性响应耦合簇理论以计算有可能的最高阶激发,并结合了选择组态相互作用(SCI),以及多n电子价态微扰理论(NEVPT2)方法。所有这些方法都与包含色散的原子基集结合使用。对于所有跃迁,作者研究了每个对称性允许跃迁的CC3/aug-cc- pvqz垂直激发能以及CC3/aug-cc- pvtz振子强度。研究表明,除了双电子激发为主的跃迁,其误差要大得多,CC3总体上给出了与较高级方法一致的激发能,典型偏差为±0.04 eV。这一工作提出了一个包含多种化合物的数据库,其中包括超过200个高度精确的激发能。研究者以其中表现最佳的理论方法为基准测试了一系列流行的激发态计算方法:CIS(D)、ADC(2)、CC2、STEOM-CCSD、EOM-CCSD、CCSDR(3)、CCSDT-3、CC3和NEVPT2,并将这些基准的结果与现有的文献数据进行了比较。
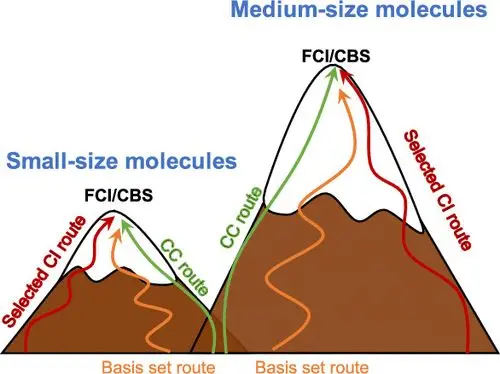
参考文献:Pierre-François Loos, Filippo Lipparini, Martial Boggio-Pasqua, Anthony Scemama, and Denis Jacquemin. A Mountaineering Strategy to Excited States: Highly Accurate Energies and Benchmarks for Medium Sized Molecules.Journal of Chemical Theory and Computation 2020 16 (3),1711-1741. DOI: 10.1021/acs.jctc.9b01216原文链接:https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b01216
3.通过梯度平方最小化来优化激发态轨道:用密度泛函理论研究单激发态和双激发态的一般方法和应用

在此,来自美国加州大学伯克利分校的Diptarka Hait&Martin Head-Gordon等人,提出了一种适用于任何量子化学轨道优化过程而不存在变分坍缩风险的一般方法。通过有限差分方法实现的最小平方梯度(SGM)方法,只需要解析能量/拉格朗日轨道梯度,且耗时仅为基态轨道优化(每次迭代)的3倍。将SGM方法应用于单行列式ΔSCF和自旋纯化的限制开壳Kohn-Sham (ROKS)方法,来研究轨道优化DFT激发态的精度。研究发现,当最大重叠法或类似的方法塌陷到基态或无法收敛时,SGM能够收敛具有挑战性的状态。研究者还报道了ΔSCF/ROKS预测了双电子激发的高度精确的激发能(这是无法通过TDDFT获得的)。通过ROKS得到的单激发态也相当准确,特别是对于对于TD-DFT颇具挑战性的Rydberg态。该结果表明,轨道优化激发态DFT方法可以突破TDDFT的限制,正确地处理双激发态、电荷转移态或Rydberg态,使它们成为研究大系统激发态的实用量子化学家工具箱中的一个有用工具。

参考文献:Diptarka Hait and Martin Head-Gordon. Excited State Orbital Optimization via Minimizing the Square of the Gradient: General Approach and Application to Singly and Doubly Excited States via Density Functional Theory. Journal of Chemical Theory and Computation 2020 16 (3), 1699-1710. DOI: 10.1021/acs.jctc.9b01127原文链接:https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b01127
4.基于神经网络实现耦合簇精度下的势能面构建:以水合质子团簇为例

高精度的势能面,是对化学系统的详细理解和预测建模的关键。为了满足这一要求,近年来研究者引进了几种基于机器学习算法、适合从头算的新型力场。在此,来自德国鲁尔波鸿大学的Christoph Schran等人,展示了如何利用高维神经网络势在耦合簇精度下自动生成有限大小簇的势能面,即CCSD(T*)-F12a/aug-cc-pVTZ。所开发的自动化过程利用已建立模型的内在属性,以无偏和有效的方式选择训练集的配置,从而最小化参考计算的计算工作量。这些思想被应用于从水合阳离子H3O+到四聚体H9O4+的质子水团簇,并得到描述所有这些系统的单个势能面,其基本收敛耦合簇的精度为0.06 kJ/mol /原子。对于四聚物的所有聚类都详细地验证了其适用性,不仅对驻点,而且对反应路径和中间构型以及不同的取样技术,都得到了可靠的结果。每一种设计,都以这种方式构建神经网络势(NNPs),以此来处理不同的情况,包括原子核的量子性质,以及覆盖极低和高温的增强采样技术。这使研究者能够快速而彻底地,探索具有聚合作用的目标质子水团簇。此外,自动化的过程,将允许处理远超出目前有限系统的情况。
参考文献:Christoph Schran, Jörg Behler, and Dominik Marx. Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground. Journal of Chemical Theory and Computation 2020 16 (1), 88-99. DOI: 10.1021/acs.jctc.9b00805原文链接:https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b00805
5.利用神经网络将多态耦合势能面表示扩展到包含性质算子:应用于氨的1,21A状态

对于可访问系统来说,使用耦合绝热状态拟合耦合绝热势能面,相比于动态动力学,非绝热动力学能够以前所未有的精度执行。动态动力学具有的优势,不仅仅体现在计算分子性质的能力,还包括电偶极矩、跃迁偶极矩和自旋轨道耦合等。这些方法的可用性,扩展了可用动态方法处理过程的范围。在此,来自新墨西哥大学的Hua Guo和美国约翰霍普金斯大学的Yafu Guan&David R. Yarkony等人,以氨的

态的电偶极矩和跃迁偶极矩的拟合为例,说明了如何利用非绝热表示法,将这些优点引入拟合耦合面的方法。

参考文献:Yafu Guan, Hua Guo, and David R. Yarkony. Extending the Representation of Multistate Coupled Potential Energy Surfaces To Include Properties Operators Using Neural Networks: Application to the 1,21A States of Ammonia. Journal of Chemical Theory and Computation 2020 16 (1), 302-313. DOI: 10.1021/acs.jctc.9b00898原文链接:https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b00898
