
2篇Nature、Cell子刊再登榜!
炎炎夏季,阻挡不了科研工作者们的前进步伐!让我们一起从科学中获得灵感,多发Paper!
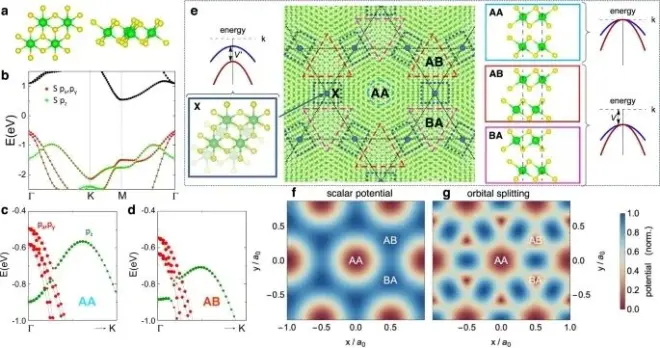
Nature Communications:扭曲ZrS2双分子层的超强自旋轨道耦合与拓扑moiré工程

在此,为了解决这个昂贵的过程,来自美国宾夕法尼亚大学的Martin Claassen & 德国马克思普朗克研究所的Dante M. Kennes
& Angel Rubio等研究者预测,1T-ZrS2的扭曲双分子层实现了一个新的可调平台,来设计由强自旋-轨道相互作用主导的二维拓扑量子相。在小的扭转角下,ZrS2异质结构产生了一个涌现和扭转控制的moiré Kagome晶格,结合几何挫折和强自旋轨道耦合,产生了一个moiré量子自旋霍尔绝缘体,具有高度可控和几乎无色散的能带。基于Γ处1T结构的价带最大值的双组分特性,研究者设计了一类过渡金属二氟族化合物的通用伪自旋理论,并研究了在拓扑moiré Kagome带的分数填充中出现的鲁棒量子反常霍尔相以及可能的分数阶Chern绝缘态。该结果建立了第IV族过渡金属二卤族双分子层作为一个新的moiré平台,在扭转可调的设置中实现强相关拓扑相。
参考文献:
Claassen, M., Xian, L., Kennes, D.M. et al. Ultra-strong spin–orbit coupling and topological moiré engineering in twisted ZrS2 bilayers. Nat Commun 13, 4915 (2022). https://doi.org/10.1038/s41467-022-31604-w
原文链接:
https://www.nature.com/articles/s41467-022-31604-w
2.Matter:机器学习辅助探索阴离子柱支撑金属有机框架用于气体分离

通过机器学习(machine learning, ML)精确预测接近真实数据的吸附性质一直是研究的方向,但由于难以获得一致、完整、准确的数据进行模型训练而阻碍了研究的进展。在此,来自浙江大学的邢华斌等研究者,将废弃的实验数据与计算数据相结合,提出了一种通用的ML模型精确预测策略,其中实验数据提供准确完整的训练数据,计算数据提供了准确一致的结构描述符。基于该开发的策略,研究者实现了对阴离子柱支撑金属有机框架中C2H2、C2H4和CO2的高精度预测。结果表明,在0.1 bar条件下,ZU-96具有较高的CO2吸附率(83.2 cm3/cm3)和较高的CO2/C2H2选择性(81.5)。在此,研究者揭示了定量的结构-性能关系,为新型吸附剂的设计提供了更直观的指导。

参考文献:
Hu, Jianbo, et al. "Machine-learning-assisted exploration of anion-pillared metal organic frameworks for gas separation." Matter (2022). https://doi.org/10.1016/j.matt.2022.07.029
原文链接:
https://www.sciencedirect.com/science/article/abs/pii/S259023852200443X#!
3.Chem:CsPbI3中γ-to-δ相变的动力学途径

全无机光活性CsPbI3钙钛矿,很容易转变为光无活性的非钙钛矿相,但其在原子水平上的转变动力学目前尚不清楚。在此,来自苏州大学的尹万健等研究者,采用基于第一性原理的随机表面行走(SSW)路径采样,来解析CsPbI3从γ到δ转变的相演化。γ(3D)→Pm(3D)→Cmcm(2D)→Pmcn(1D)→δ(1D)的最低能量路径具有低至~31 meV/原子的跃迁势垒。γ-pm过渡被确定为性能控制步骤。此外,根据鲍林定律和结合能,得到了由取代掺杂离子大小决定的火山型过渡势垒。相变过程中[010]面应变变化最大,说明涉及[010]轴的应变对增加相变势垒的影响更显著。这些结果为制备稳定、长期的全无机卤化物CsPbI3钙钛矿,提供了合理的建议和指导。

参考文献:
Chen, Gao-Yuan, et al. "Kinetic pathway of γ-to-δ phase transition in CsPbI3." Chem (2022). https://doi.org/10.1016/j.chempr.2022.07.026
原文链接:
https://www.sciencedirect.com/science/article/abs/pii/S2451929422003850#!
4.JACS:多水平计算研究揭示了轴向配体在Fe-N-C材料氧还原反应中的重要性

由于对电化学条件下氧还原反应(ORR)的不完全原子理解,燃料电池应用的Fe-N-C材料的系统改进,已被证明是具有挑战性的。在此,来自美国西北国家实验室的Simone Raugei & 美国耶鲁大学的Sharon Hammes-Schiffer等研究者,采用一种结合从头算分子动力学模拟和恒势密度泛函理论计算的多级计算方法,评估了关键ORR中间体的质子耦合电子转移(PCET)过程和吸附热力学。这些计算表明,Fe-N-C材料ORR的电位限制步骤是FeIII-OOH中间体的形成。他们还表明,在整个催化循环中,水分子轴向连接到铁中心的活性位点模型产生的结果与实验测量结果一致。特别是,要可靠地预测ORR起始电位和Fe(III/II)氧化还原电位(与FeII-OH转化为FeII和解吸的H2O有关),需要有一个轴向共吸附在铁中心的H2O。五坐标而不是四坐标活性位点的观测,对ORR的热力学和机理具有重要意义。这些发现强调了在现实的电化学条件下,溶剂-基底相互作用和表面电荷效应,对理解PCET反应机制和过渡金属氧化还原偶联的重要性。

参考文献:
Hutchison, Phillips, et al. "Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe-N-C Materials." Journal of the American Chemical Society (2022). https://doi.org/10.1021/jacs.2c05779
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.2c05779
5.Nat. Comput. Sci.:非绝热分子动力学模拟中半导体非平衡载流子的有效寿命

用非绝热分子动力学计算的半导体中非平衡载流子的寿命,往往与实验结果相差数量级。在此,来自复旦大学的陈时友等研究者,通过重新讨论了载流子寿命的定义,报告了一个系统的程序来计算半导体晶体在现实条件下的有效载流子寿命。考虑所有的复合机制和使用适当的载流子和缺陷密度,对于弥合建模和测量之间的差距至关重要。研究者计算的CH3NH3PbI3的有效载流子寿命与实验结果一致,但受能带间辐射复合和肖克利-读-霍尔缺陷辅助非辐射复合的限制,而能带间非辐射复合可以忽略不计。该方法通过在化合物半导体CdTe和GaAs上的应用进一步验证,从而可以应用于其他材料系统的载流子寿命模拟。

参考文献:
Wang, S., Huang, M., Wu, YN. et al. Effective lifetime of non-equilibrium carriers in semiconductors from non-adiabatic molecular dynamics simulations. Nat Comput Sci 2, 486–493 (2022). https://doi.org/10.1038/s43588-022-00297-y
原文链接:
https://www.nature.com/articles/s43588-022-00297-y
