
【菜鸟博士学习】MSConvertGUI+seeMS导出质谱离子峰进行分析
MSConvertGUI+seeMS导出质谱离子峰进行分析
MSConvertGUI + seeMS induced mass spectrometry ion peak for analysis
代谢组学(metabolomics) 是在后基因组学时代兴起的一门跨领域学科, 其主要目标是定量的研究生命体对外界刺激、 病理生理变化、 以及本身基因突变,而产生体内代谢物水平的多元动态反应。 代谢组学分析可分为非靶向代谢组学(Untargeted metabolomics) 和靶向代谢组学(Targeted metabolomics) 。
Metabolomics is a cross-disciplinary discipline emerging in the post-genomics era, whose main objective is to quantitatively study the external stimuli, pathophysiological changes, and genetic mutations of living organisms, but produces in vivo metabolite level multivariate dynamic response. Metabonomics analysis can be divided into Untargeted metabolomics and Targeted metabolomics.

非靶向代谢组学是尽可能多地定性和相对定量生物体系中的代谢物, 最大限度反映总的代谢物信息。根据这一目的, 非靶向代谢组学常采用高分辨质谱仪,如飞行时间质谱、 四极杆-飞行时间质谱、 Orbitrap 质谱及 FT-ICR 质谱等; 有利于检测出尽可能多的代谢物和结构鉴定。然而目前非靶向代谢组学方法在定量准确性、 代谢物鉴定和后期数据处理等方面都存在着一定的局限性。 特别是对于原始离子峰的提取和分析,今天科研小汪给大家介绍一个可以将任何质谱仪器采集的原始LC-MS数据直接一键式提取所有离子峰的方法,并进行简要分析!~
Non-targeted metabonomics is to characterize and quantify as many metabolites as possible in biological systems and to reflect the total metabolite information to the maximum extent. For this purpose, non-target metabonomics is usually characterized by high resolution mass spectrometry, such as time-of-flight mass spectrometry, quadrupole-time-of-flight mass spectrometry, orbital rap mass spectrometry and FT-ICR mass spectrometry It is helpful to detect as many metabolites as possible and identify their structures. However, the current non-targeted metabonomics methods have some limitations in quantitative accuracy, metabolites identification and late data processing. In particular, for the extraction and analysis of the original ion peaks, today scientific researcher Xiaowang introduced a LC-MS method that can be used to directly extract all the ion peaks from the original LC-MS data collected by any mass spectrometer, and a brief analysis! ~
1、原始数据格式转换。
1. Raw data format conversion.
现在质谱仪器有很多,但是各个仪器都有自己的采集模式和分析软件,产生的数据格式也不一样,往往一些专业的代谢组学分析网站(Xcms or metlin)或者软件都要求特定的数据格式,为了便于分析,我们先将这些数据转换成统一格式。格式转换软件有很多,我给大家推荐的是ProteoWizard中的MSConvertGUI。这个软件很好用,直接将数据导入后(Browse),选择输出格式(Output form), 一般为mzXML居多。然后点击Start即可。
Now there are many mass spectrometer instruments, but each instrument has its own collection mode and analysis software, and the data format produced is also different, some professional metabonomics analysis web site (Xcms or metlin) or software requires specific data format, in order to facilitate analysis, we will first convert the data into a unified format. Format conversion software there are many, I would like to recommend to you is the proteizard msconvert Gui. This software is easy to use, directly after the data import (Browse) , choose Output form, generally mzXML in the majority. Then click Start.
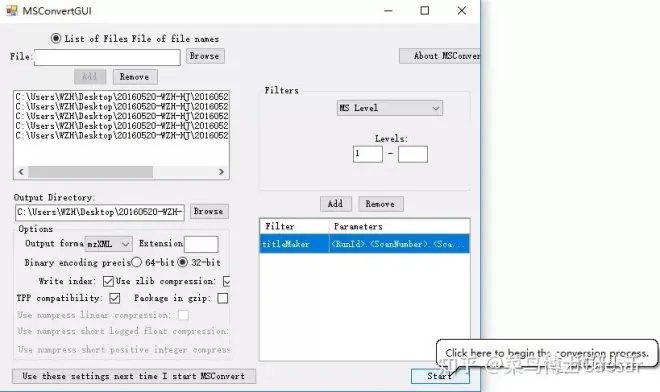
2、将转换的格式导入seeMS软件提取离子峰(也可以导入Xcms等网站在线搜索)。
Import the converted format into the seeMS software to extract the ion peaks (you can also import online searches such as Xcms) .


如图所示,用seeMS打开mzXML格式数据后,即可看到软件提取的所有离子峰,包括保留时间,分子量,丰度值等
As you can see in figure, when you open up the data in the seeMS MZXML format, you can see all the ion peaks extracted by the software, including retention time, molecular weight, abundance, and so on

3、将seeMS软件提取的离子峰导出到EXCEL并进行分析。
3. Export the ionic peaks extracted by the seeMS software to EXCEL and analyze them.
我们将每组提取到的m/z,RT及intensity等信息复制到excel即可进行差异倍数计算及PCA分析等(具体数据分析见EXCEL数据处理)。
The information of M/Z, RT and intensity extracted from each group is copied to EXCEL, and then the difference multiple and PCA can be calculated.


4、找出差异性成分并分析结果。
4. Find out what’s different and analyze the results.
我们将差异性成分再投到相应软件及网站进行火山图、热图等绘制,并进一步进行pathway等分析(具体代谢组学数据分析)
We put the differential components into the corresponding software and website for volcano mapping, thermal mapping, and further pathway analysis (specific metabolomic data analysis)



