
金属参与的有机反应-醇到羰基化合物的氧化反应
旧文重发,转自我自己的微信公众号
金属或类金属试剂在有机化学中参与多种类型的反应,其中使用金属铬氧化剂实现从醇到羰基化合物的氧化反应是有机化学里最基础也是最常见的反应,在药物研发,天然产物合成中几乎都少不了一步对含有羟基官能团的氧化。本系列文章将以某一个金属和特定的反应为载体,介绍一些十分经典,常用的试剂以及所涉及的基础理论。
铬类试剂:
1.1 介绍:
三氧化铬是一种深红色吸湿性的晶体,具有强氧化性。将其溶解在水中将形成铬酸溶液并与多种聚合体呈平衡状态。

1.1.1. Jones 试剂
尽管三氧化铬在某些有机溶剂如叔丁醇,吡啶和醋酸酐中可溶,它在这些溶剂中由于具有爆炸性而应用性受限。虽然如此,可以将丙酮与三氧化铬的稀硫酸溶液安全的混合。这也就是Jones氧化所采用的基本原理:将三氧化铬的稀硫酸溶液滴加到含有所需氧化的有机物的丙酮溶液中。这个手段和反应第一次由Jones报道,也变成了将醇氧化为羰基化合物最常用的反应之一,为日后铬氧化试剂的发展奠定了基础。
醇被铬类试剂氧化的一般性机理可以这样描述:醇1与体系中的铬物种快速形成铬酸酯2,铬酸酯分解(决速步)得到氧化产物。

如果第一步氧化得到的是醛,如果体系中存在水,且该醛的活性足够,则可以继续形成水合物4并进而被氧化为酸5。
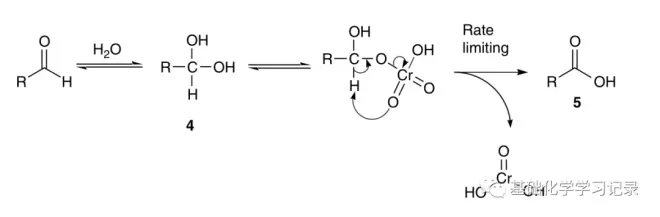
我们已经知道上述机理中铬酸酯的形成一步是快速的,这样会导致一个有趣的现象:铬类试剂往往对于空间位阻相对较大的醇类反应物比空间位阻相对较小的类似底物表现出更高的活性,这与我们一般的直觉完全相反。这是因为铬酸酯中间体的交换是快速的,即便是位阻很大的位置也较其决速步快得多,但是在位阻大的地方形成的铬酸酯在分解成羰基产物的时候可以伴随着更多的张力缓解,这对铬酸酯的分解有着明显的促进效应。因此一个很直观的例子就是:在环状底物中,处于直立键位置的羟基较处于平浮键位置的羟基氧化速率更快,因为直立键的羟基受到的1,3-二直立键相互作用在形成氧化产物羰基之后可以被释放进而促进了直立键铬酸酯的分解。
尽管Jones氧化剂可以高效的将二级醇转化为酮类化合物,但是其氧化一级醇得到醛的产率往往不高,而是发生过度氧化得到酸,仅有当醛的水合物生成不利的情况下才能高产率的得到醛。
1.1.2. Sarett以及Collins试剂
三氧化铬与吡啶的反应伴随着强烈的放热,形成复合物CrO3-2Py,在许多有机溶剂中可溶。将其溶解在吡啶中形成的溶液被称为Sarett试剂。由于其中没有水或很少有水,其不仅能将二级醇氧化为酮,还能将一级醇氧化为醛。对其的一个改进是将CrO3-2Py溶解在CH2Cl2中,这被称为Collins试剂。该试剂较Sarett试剂有着更多的优点,比如CH2Cl2较吡啶来说碱性明显较弱,可以避免因为吡啶的碱性带来的不必要麻烦。
需要注意的是在形成CrO3-2Py的过程中有一定的危险,一定要将CrO3加到吡啶当中,二者顺序不能颠倒否则容易发生爆炸。CrO3-2Py高度吸湿,在存在有机物的时候容易爆炸,可以通过在CH2Cl2中原位形成CrO3-2Py的策略避免爆炸的危险,具体操作便是将三氧化铬加到搅拌中的吡啶与CH2Cl2混合溶液当中,这也几乎是现在使用CrO3-2Py作氧化剂时唯一采用的手段。在操作含三氧化铬的试剂时务必按照已有的文献进行操作,严禁私自篡改或“改进”操作程序。CH2Cl2是最常用的溶剂因为它十分耐火,严禁在没有相关文献的前提下私自更换溶剂进行实验。
1.1.3. 重铬酸二吡啶鎓盐 (Pyridinium Dichromate-PDC)
当将吡啶加到三氧化铬的水溶液当中时便可能获得PDC沉淀。

这是一个亮黄色的固体,在许多有机溶剂中可溶,储存和处理都很方便,因为它不吸湿。PDC一般于室温下在CH2Cl2中使用,能高效的将一级醇氧化为醛,二级醇氧化为酮。由于此体系中缺乏水,因此可以得到醛类产物。
1.1.4. 氯铬酸吡啶鎓盐 (Pyeidinium Chlorochromate-PCC)
CrO3与盐酸作用导致一个平衡反应见下图,当加入吡啶时立刻得到PCC沉淀。

该试剂为橘黄色固体,与PDC有许多共性,二者几乎可以互相代替。
1.1.5. 氧化剂的选择
下面列出几条在特定条件下如何选择氧化剂的条款。
Jones试剂很容易操作,因为不涉及任何无水条件,此外它很便宜。在某些耐酸的底物中可以进行高量级反应。但一般不能高效的得到醛。
Collins试剂也很便宜,但需要严格的无水条件。尽管有时候它缺乏像PDC或PCC那样的选择性,在涉及某些不复杂的底物参与的反应中它可以高效的得到醛和酮。
PDC和PCC较贵但往往给出最佳的反应结果。
1.2. Jones 氧化
该氧化涉及CrO3,H2SO4,底物和水,氧化剂本身可看作是铬酸或其衍生物,因此用Na2Cr2O7或K2Cr2O7代替CrO3也可以得到相同的结果。该反应操作简单,无需特殊条件多用于将二级醇氧化为酮,不用于或很少用于得到醛产物。还需要注意的一点是该体系酸性较强,有时不适用于对酸敏感的底物。但需要说明的是,该体系其实涉及两相-有机相与水相,因此很多时候水相中的酸对有机相里的底物影响并不是很大,很多对酸敏感的保护基在该条件下并没有被脱除,此外也可以通过降低硫酸的浓度来进一步达到防止脱除保护基的目的,尽管这会降低Jones试剂的氧化能力。下面是一些代表性反应并附有简单的说明。
实现了克量级不稳定酮类化合物的制备

酸敏感的缩酮和缩醛基团均没有被脱除

叔丁基酯在该反应中没有被脱除

同时实现了烯丙醇,半缩醛,醛基的氧化

可能是由于质子化的原因易被氧化的氮均没有被影响

这个很困难的氧化反应使用Swern,Collins,MnO2,TEMPO,PCC以及DMP均失败了,只有Jones试剂在低温下才得到了满意的结果,产物中的一个羰基互变异构为对氧化剂十分敏感的烯醇结构。

1.2.2. 保护基与官能团对Jones氧化的敏感性
尽管Jones氧化条件含有水相硫酸,对酸敏感的官能团或保护基由于水相和有机相的分离往往可以不被影响。只有那些对酸极度敏感的保护基才在该条件下被脱除,如果脱除保护基后暴露出新的羟基官能团,那么它会被原位氧化。
需要说明的是,在不同底物参与的Jones氧化中常常使用不同的酸度,温度以及反应时间,一个特定的保护基或官能团对酸的敏感度也会受到这些因素的影响。下面描述的只是一些经验规则,具有一定的相对性。
大多数的硅醚保护基都可以在该条件下被保存,除了对酸十分敏感的TMS之外。
对于醚类保护基而言除了THP醚之外,醚类保护基均在该条件下被保存。
苄基,PMB,叔丁酯之类的保护基不受影响,但三苯甲基及其衍生物对酸太过于敏感,不能经受这类反应。
醛一般会被氧化为酸,半缩醛会被氧化为内酯,含硫官能团被氧化为亚砜或砜,除了能形成很稳定的碳正离子的环氧化物以外,环氧化物不受该反应的影响,胺类,吡啶,酯也不受影响。
1.2.4. 原位脱保护-氧化
一些对酸十分敏感的保护基被脱除的同时会暴露出一个羟基或易被氧化的其他官能团,在这样的情况下会发生一锅脱保护-氧化反应。
THP醚脱除并氧化

TMS硅醚脱除并氧化,TBDPS硅醚未被影响

TBS硅醚较为稳定,但可以在外加的HF或KF的情况下在Jones氧化反应下被脱除并氧化。

1.2.5. 通过Jones氧化得到醛
如前文所述由于醛水合物的形成,在Jones氧化下往往会得到酸。但如果考虑到水合物的占比在某些情况下是十分少的甚至可以忽略不计,高产率的得到醛似乎也是可以实现的。通过供电子基团,共轭效应,立体位阻来降低醛的活性从而降低水合物平衡占比或将生成的醛通过不断的蒸馏而移除反应体系等手段在一定的条件下确实可以高产率的得到醛,但这也仅仅是在底物不复杂的情况下才行。用甲乙酮代替丙酮作溶剂可以有效降低醛的水合程度,因为甲乙酮的极性小于丙酮。
1.2.6. 副反应
一些带有能形成稳定碳正离子取代基的羟基化合物在进行反应的时候除了正常的断裂碳氢键外还可能会伴随着碳碳键的断裂而得到复杂的产物。

下面是一些实例。
在该例中碳碳键断裂可以得到较为稳定的三级碳正离子因此副反应明显了很多

在三级醇羟基的孤电子的推动下导致中间环的裂解反应成了主反应

该例与前者类似但还伴随着桥环张力的释放作为推动力

这是一个十分有趣的例子将在下面详细讨论

下面是一种可能的机理,三级碳正离子的稳定性以及环张力的释放推动着铬酯的分解,形成的碳正离子被分子内的醛基捕获进而水合为半缩醛,半缩醛被氧化得到最终产物。

需要说明的是在涉及含有小环的醇参与的铬(VI)试剂氧化时,反应过程中生成的Cr(IV)会开启自由基路径(详情请参考J.Am.Chem.Soc,1973,7123-Mechanism of the Chromic Acid Oxidation of Cyclobutanol-简略的概述一下类似文献的内容:Cr(VI)对于醇的氧化一般为两电子转移先得到Cr(IV)但是在特殊底物如羟基乙酸做底物时可能生成底物与铬的2:1配合物发生一步3电子转移直接得到Cr(III),Cr(IV)活性很高,是高效的单电子氧化剂,可以将有机物氧化为自由基并进而引发碳碳键断裂反应,自身变为Cr(III),自由基被Cr(VI)或Cr(IV)氧化为正常的氧化产物或碳碳键断裂的产物,其中Cr(VI)产生Cr(V),这是一个相对不活泼的中间体,参与双电子氧化过程得到Cr(III)),因此我们来考虑下面的自由基机理,由于底物涉及多环估计,最初形成的自由基应该不会改变其构型也就是自由基是占据直立键位置的,因此氧化剂从直立键位置给出羟基,进而闭环得到顺式并环结构。

实验证明,相同的底物用PCC或PDC氧化只能得到四元环未被打开的酮产物,可能是因为体系中的吡啶的存在使得铬氧化剂的氧化能力明显下降导致的结果。但是将该产物用Jones氧化仍然得到了5元环内酯,说明下述机理也可能存在:经历一步类似于BV重排的二电子三元环过渡态,按照分子轨道对称性守恒原理,这个过程应当是“同面的”因此左方的手性碳构型保持,这与产物的立体化学结果完全一致。

在这个例子中可能由于氮原子被质子化而无法协助碳碳键断裂

下面的例子中没有得到由醚氧原子推动导致碳碳键断裂的产物的原因可能是旁边的羰基对形成的碳正离子的去稳定化效应。

下面两个例子说明即便符合碳碳键断裂的前提条件,一般来说碳氢键断裂活化能更低,在温和的条件下仍然以碳氢键断裂为主反应。

有时候,羟基形成的铬酯在空间位置合适的位置发会诱导发生环氧化反应。如下图所示,直立键的羟基形成的铬酯由于空间位置合适,且附近有一个双键,因此发生了分子内环氧化反应。

换用平浮键羟基异构体则由于形成的铬酯的空间位置不合适而没有发生环氧化反应。这说明该反应对立体电子的需求极其严苛。

有时,通过氧化新形成的羰基会进一步诱导生成不饱和羰基化合物。这对底物的特殊结构要求很高。

三级醇附近合适的位置如果存在双键,则中间体铬酯可能会先发生[3,3]迁移再进行氧化。

上面的迁移反应与普通的氧化反应相比明显较慢,因此可以实现选择性的氧化二级醇。

有时烯丙基二级醇形成铬酯也会发生迁移反应并伴随着环氧化反应得到复杂产物。再一次的这种情况只发生在直立键取代的醇上,迁移与不迁移的比率为3:5.

1.3. 其他含铬氧化
铬类氧化剂有许多通性,其细节已经在前文中详细叙述,这里只讨论它们一些其他的特殊应用。
由于吡啶配位于金属铬上导致其氧化能力降低,因此该类反应较Jones氧化温和的多,可以获得高产率的醛。该类试剂也可以将烯丙位或苄位氧化得到羰基化合物,但这样的过程比一般的醇氧化慢的多,可以在双键存在的情况下实现醇的选择性氧化。许多保护基和易被氧化的官能团如硫代缩醛都能在该反应条件下被保存,其中二级醇的TMS保护基不被影响,但一级醇的TMS保护基会被脱除,因此可以实现下图的在硫代缩醛和多个二级醇存在下选择性氧化一级醇。

含硒官能团会被氧化为硒亚砜,半缩酮会以其开环形式被氧化,但该氧化过程比一般的醇氧化慢,可以实现在半缩酮结构中选择性氧化普通的醇羟基。

由于铬类试剂的氧化机理是类似的,因此前面所述的一些副反应同样会出现在该情况下。

有时,底物的羟基可以诱导分子内的跨环方式对烯烃的氧化,立体选择性的生成特殊的四氢呋喃环系。

PCC中铬部分带负电荷,可以认为具有一定的亲核性,如KMnO4一样。在不是很强的酸性条件下以SN2反应打开环氧环,见下图。

1,2-二醇在某些条件下可以在铬类试剂存在下发生氧化切断碳碳键的反应,这类反应最用的是碘(VII)和铅(IV)试剂,它们也是最高效的,铬类试剂一般不高效,因为其机理不像前二者一样可以走环状协同步骤,由于铬的配位倾向性与碘(VII)和铅(IV)不同且在铬在被还原时电子留入铬的d轨道,而铅和碘试剂被还原时电子则留入p轨道,由于d轨道和p轨道的对称性存在明显差异,因此这两类试剂的环状过渡态的对称性允许性也不同。如下图所示,6电子的环状过渡态对于碘来说是芳香性的,而对于铬参与的轨道来说由于存在一个截面因此是反芳香性的。(参考Tetrahedron 1971, 27, 81)

结果就是1,2-二醇形成的环状铬酯的分解只能是分布的而不能是协同的,因此有较大的势垒,这也是为何铬试剂一般不用于1,2-二醇的切断。
1,4-二醇如果空间位置合适则形成内酯(但也有不形成内酯的情况),不合适则就被分别氧化。1,5-二醇类似。


烯丙基正离子的稳定性和桥环张力的释放促使了下面的重排反应的发生。

铬酯也算是一个好的离去基团,因此在某些特定的情况下会得到像取代或消除反应一样的产物。当然要想得到取代为主的产物必然要求某些特殊的结构因素,见下面这些例子。




下面是一个消除-环氧串联的例子。

总结:
本文列举了大量的铬试剂参与的醇氧化反应,着重介绍了其涉及的特殊反应,算是一个对金属铬参与的醇氧化反应的综述。
