
Gromacs: 蛋白-配体复合物实例 (1)
为了更好地对gromacs进行理解和学习,在这里将借gromacs官方实例的练习进行一个记录。
蛋白配体复合物在gromacs中的步骤可以总结成:
生成拓扑文件 ————> 定义盒子与溶剂 ————> 加入离子(目的是让体系的净电荷为0) ————> 能量最小化 ————> NVT, NPT平衡 ————> MD ————> 分析。
其步骤和之前基础的溶菌酶在水体系的平衡相差不大,之后将用官方实例作一次练习。
在本次实例中,将使用T4溶菌酶与JZ4配体进行动力学模拟
步骤一 : 准备蛋白拓扑
首先去RCSB下载T4溶菌酶的pdb文件(PDB: 3HTB)

在pymol中,T4溶菌酶的结构显示为这样

接下来,需要将对T4溶菌酶去除结晶水
grep -v HOH 3HTB.pdb > 3HTB_clean.pdb
在准备好T4溶菌酶后,我们现在面临的问题是,JZ4 配体在 GROMACS 提供的任何力场中都不是可识别的实体,因此如果您尝试将此文件传递给 pdb2gmx,它将给出致命错误。仅当力场的 .rtp(剩余拓扑)文件中存在构建块条目时,才能自动组装拓扑。由于情况并非如此,我们将分两步准备系统拓扑:
1. 使用pdb2gmx准备蛋白质拓扑
2. 使用外部工具准备配体拓扑
由于我们将分别准备这两种拓扑结构,因此必须将蛋白质和 JZ4 配体分别保存到不同的坐标文件中。像这样保存 JZ4 坐标。
grep JZ4 3HTB_clean.pdb > jz4.pdb
得到的JZ4长这样

然后只需删除 3HTB_clean.pdb 中的 JZ4 即可。至此,准备蛋白质拓扑结构就变得轻而易举了。本教程将使用的力场是 CHARMM36,可从 MacKerell 实验室网站获取。在那里下载最新的 CHARMM36 力场压缩包和 "cgenff_charmm2gmx.py "转换脚本,我们稍后将使用该脚本。下载与您安装的 Python 版本(Python 2.x 或 3.x)相对应的转换脚本版本。
在下载完力场文件后在工作目录中解压力场压缩包:

现在您的工作目录中应该有一个 "charmm36-jul2022.ff "子目录。用 pdb2gmx 生成一个 T4 溶菌酶的拓扑结构:
gmx pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro -ter
系统将提示你进行 3 项选择。
1. 力场
2. 水模型
3. 终点类型
本教程选择 CHARMM36 force field(选项 1),它列在 "从当前目录 "列表的第一位。
选择默认水模型(CHARMM-modified TIP3P),然后为末端选择 "NH3+"和 "COO-"。由于 N 端残基为蛋氨酸 (MET),pdb2gmx 会选择与碳水化合物不兼容的末端类型,因此这种交互式选择是必要的。您必须选择特定于蛋白质的末端,否则会出现原子名称不匹配的致命错误。

这里我出现了个PO4的错误,原因是没有删除PO4这个小配体,在实际操作中要将其他配体删干净.
步骤二:准备配体拓扑
我们现在必须处理配体问题。但是,对于力场无法自动识别的某些种类,我们该如何为其提供参数呢?正确处理配体是分子模拟中最具挑战性的任务之一。力场作者花费数年时间推导出自洽的力场,而在此框架中引入一个新种类并非易事。任何新种类的力场参数都必须以与原始力场一致的方式推导和验证。
对于 OPLS、AMBER 和 CHARMM 力场,这种推导通常采用各种量子力学计算的形式。这些力场的主要文献介绍了所需的程序。对于 GROMOS 力场,参数化方法并不明确,主要依赖于凝聚相行为的经验拟合。也就是说,为每种原子类型计算一些初始电荷和伦纳德-琼斯参数,评估其准确性并加以改进。虽然最终结果非常令人满意,即流体与现实世界中的流体非常相似,但推导过程可能会非常费力。
因此,自动化工具非常受欢迎。对于每种力场,都有一些方法或软件程序声称可以提供与各种力场兼容的参数。但并非所有的方法或软件都同样准确。请参考下表中的几个例子:

为 JZ4 添加氢原子
在本教程中,我们将使用 CGenFF 服务器生成 JZ4 拓扑。在访问网站后注册账户并激活。CGenFF 需要 Sybyl .mol2 文件作为输入,以收集基本的原子类型信息和键合连接性。CHARMM 也是一种全原子力场,即明确表示所有 H 原子。晶体结构通常不会指定 H 原子坐标,因此必须将其内置。要生成 .mol2 文件并添加 H 原子,使用 Avogadro 程序。在 Avogadro 中打开 jz4.pdb,从 "Build(构建)"菜单中选择 "Add Hydrogens(添加氢原子)"。Avogadro 将在 JZ4 配体上构建所有氢原子。保存名为 "jz4.mol2 "的 .mol2 文件(文件 -> 另存为...,从下拉菜单中选择 Sybyl Mol2)。
以上是官方的表述,在这里,我使用openbabel对配体进行加氢。
obabel -ipdb jz4.pdb -omol2 -Ojz4.mol2 -h
使用 CGenFF 生成 JZ4 拓扑
现在,jz4.mol2 文件已可用于生成拓扑结构。访问 CGenFF,登录账户,然后点击页面顶部的 "Upload molecule"。上传 jz4.mol2,CGenFF 服务器将以 CHARMM "stream"文件(扩展名为 .str)的形式快速返回拓扑图。将其内容从网站保存到名为 "jz4.str "的文件中。
CHARMM stream文件包含所有拓扑信息 - atom类型、 charges和 bonded连通性。它还包含额外的结合参数,这些参数是通过类比力场未涵盖的内部相互作用而生成的。CGenFF 还为每个参数提供了惩罚分数,即评估所分配参数的可靠程度。低于 10 的值可直接使用。10 - 50 之间的值意味着需要对拓扑结构进行一些验证,而大于 50 的惩罚值通常需要手动重新参数化。这种惩罚性评分是 CGenFF 服务器最重要的功能之一。许多其他服务器生成的拓扑结构都是 "黑盒子",用户只能默默信任。将整个研究项目寄托在未经验证的自动程序上是非常危险的。糟糕的配体拓扑会导致大量时间的浪费和不可靠的结果。请务必验证新参数化的拓扑结构!至少要根据力场中的现有分子检查配体的电荷量和原子类型。
检查 jz4.str 的内容,查看电荷和新二面参数的惩罚。所有惩罚值都非常低,表明该拓扑结构质量非常好,可以直接用于我们的模拟。
CHARMM 格式的 JZ4 拓扑很好,但如果我们要在 GROMACS 中运行,它就没用了。请使用我们之前从 MacKerell 网站下载的 cgenff_charmm2gmx.py 脚本。脚本名称中会包含版本 _py2 或 _py3,但为简单起见,此处省略了这些信息,但您需要使用下载的 Python 版本脚本的实际名称。使用以下命令执行转换。
python cgenff_charmm2gmx_py3_nx2.py JZ4 jz4.mol2 jz4.str charmm36-jul2022.ff
在运行这个程序的时候,在我目前的python3.9的环境中会出现一些错误,需要对脚本进行针对性的修改才能跑通。要注意环境的问题。
跑完显示:

该配体引入了不属于现有力场的新键合参数,这些参数被写入一个名为 "jz4.prm "的文件,其格式为 GROMACS .itp 文件。我们稍后将处理该文件,但必须注意到它的存在。现在配体拓扑结构被写入 "jz4.itp",其中包含配体[ moleculetype ] 定义。
通过 pdb2gmx,我们得到了一个名为 "3HTB_processed.gro "的文件,其中包含经过处理的、符合力场的蛋白质结构。我们还有来自 cgenff_charmm2gmx.py 的 "jz4_ini.pdb "文件,其中包含所有必要的 H 原子,并与 JZ4 拓扑中的原子名称相匹配。使用 editconf 将此 .pdb 文件转换为 .gro 格式:
gmx editconf -f jz4_ini.pdb -o jz4.gro
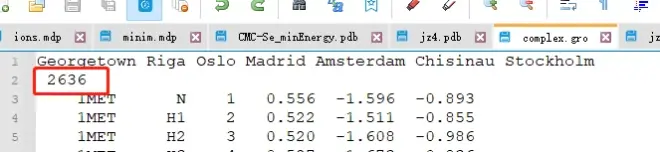
将 3HTB_processed.gro 复制到一个新的待处理文件中,例如将其命名为 "complex.gro",因为将 JZ4 添加到蛋白质中将生成蛋白质配体复合物。接下来,只需复制 jz4.gro 中的坐标部分并将其粘贴到 complex.gro 中,位置在最后一行蛋白质原子的下方和方框矢量之前,就像这样:


由于我们已在 .gro 文件中添加了 22 个原子,因此请增加 complex.gro 的第二行以反映此更改。现在坐标文件中应该有 2636 个原子。
构建拓扑
在系统拓扑中加入 JZ4 配体的参数非常简单。只需在包含位置限制文件后,在 topol.top 中插入一行 #include "jz4.itp"即可。位置限制文件的包含表示 "蛋白质 "分子类型部分的结束。


配体引入了新的二面性参数,这些参数由 cgenff_charmm2gmx.py 脚本写入 "jz4.prm"。在 topol.top 顶部插入 #include 语句,添加这些参数:

这个 #include 语句的位置非常重要 ‘-’ 必须出现在任何 [ moleculetype ] 条目之前,因为在构建任何分子之前必须先定义所有参数。它还必须出现在父力场的 #include 语句之后,因为在引入使用原子类型的键合参数之前,必须先了解所有原子类型。
最后要进行的调整是在 [ molecular ] 指令中。考虑到complex.gro中有一个新分子,我们必须将其添加到此处,如下所示:

现在,拓扑结构和坐标文件与系统内容一致。
下一节:添加溶剂
