
荧光定量(qPCR)原理、实验流程及结果分析

一、原理&应用
一)PCR

二)qPCR——化学原理


1.SYBR Green

注:具有结合双链小沟的结构特性,体系中有非特异性扩增时也是可以结合的,所以要保证产物是特异性的
2.TaqMan Probe

TaqMan Probe:20nt(碱基)的寡聚核苷酸,两端有5-发光基团,3-淬灭基团,探针完整时二者距离近不发光。dna外切酶可水解探针,距离变远发光。
注:探针的退火温度要比引物高5-8度。原因:退火时要让探针比引物先结合到模板上,才能被外切酶水解
3.SYBR 对比 TaqMan Probe

三)qPCR——数学原理

no:初始模板量,n:循环次数,N:终产物数量,e:扩增效率;Ct:循环数;k/b:常数
注:实验过程有消耗,实际方程为红色那个
计数方法:仪器给出CT值,带入线性方程计算初始浓度N0
四)应用

二、流程&注意事项
一)流程

1.RNA提取
1)RNA

RNA易降解原因:(1)单链(2)核糖核酸上的羟基(3)RNAase存在于周围环境,反应剧烈
提取原理:(1)尽量不要使RNA降解
2)流程


图:动物细胞/动物组织/植物细胞/血液

- 动物组织更推荐液氮研磨,匀浆会温度升高/遇到内外源性RNAase
- 植物组织有细胞壁更推荐液氮研磨
- 细胞样品简单只需得到细胞沉淀

β-巯基乙醇可变性蛋白释放核酸

Trizol法——沉淀法(传统经典)
胍盐——过柱法



完整RNA三条带:20、88、5,第一条带亮度是第二条的两倍,第三条最浅
中间图条带二最亮——表明RNA部分降解
右边图条带三最亮——表明RNA严重降解,需重新提取RNA
提取关键点

2.逆转录
1)逆转录反应

此过程会加入逆转录酶、引物、BUffer
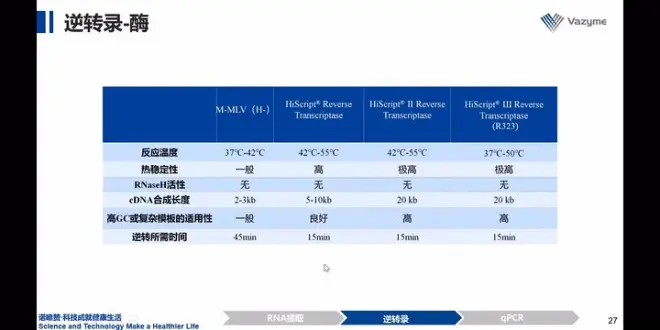
2)逆转录酶

两种常用酶:M-MLV/AMV均具有RNAase的活性,降低cDNA的完整性,当目的片段十几kb不能选择此酶,更换下表二肽酶/三肽酶
3)逆转录引物

三大类:
1.Oligo dT/锚定Oligo dT
2.随机引物:6个随机碱基的结合,原核生物RNA无poly A尾不能用.Oligo dT/锚定Oligo dT
3.基因特异性引物

原因:在MAP4基因5‘/3’分别设计引物,单独使用Oligo dT得到的CT值有差异(上图),单独使用逆转录出来的cDNA有严重的3‘端偏好,建议使用Oligo dT和随机引物的混合引物
4)金属残留

电泳图单条带上方有一个较大的片段,为金属DNA残留
右图:
绿色:未去除DNA逆转录出
红色:未去除未逆转录,即RNA,不能作为qPCR模板,出现曲线的原因:残留的DNA引起的有污染曲线
黄色:去除DNA逆转录(正确)
黄绿相差3个CT值,建议去除DNA组干扰
3.qPCR

100-200bp时扩增效率才能90%—100%

验证引物的预实验:
将模板梯度稀释——用此引物做pcr——1.熔解曲线:如图窄窄的单峰=扩增特异性强
稀释倍数与CT值做标曲,求斜率带入计算扩增效率的公式

查文献看别人用什么/网站

1.不引入内参:在两组细胞中,提取RNA——逆转录——qpcr——得到CT值和ΔCT——代入公式计算
此实验不严谨:∵保证取RNA——逆转录——qpcr的效率均为一致
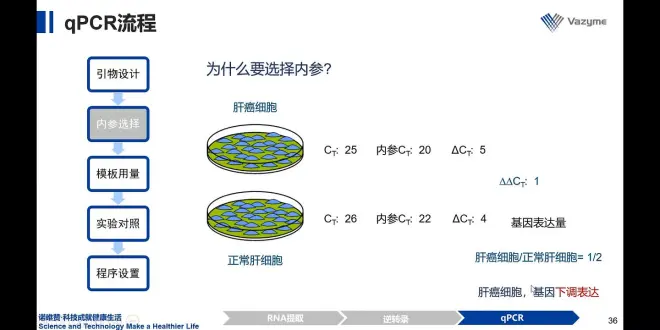
2.引入内参:结果相反


先进行预实验


三、结果分析
一)常见图谱&名词
1.校正染料

2.扩增曲线

- 基线期:荧光背景阶段
- 指数增长期:着重关注,方程(n≈n0)此时才适用
- 线性增长期:酶/dNTP不断消耗
- 平台期:


阈值:最低荧光下线的荧光信号

阈值线决定CT值大小
3.熔解曲线

左图解释:PCR反应完成后,仪器升温(60—95℃),此时双链DNA开始解链,荧光信号下降,至某一数值陡降,此时的温度为Tm。
右图:每个锋代表特异性产物,要求溶解曲线单峰,出现杂峰意味着有非特异性的扩增
4.CT值有效性?

1.溶解曲线单峰。如果不是单峰,有非特异性扩增的污染导致曲线上升。

2.复孔间重复性不好

右下粉色图:NTC熔解曲线在目的样本峰形图前有→认为是引物二聚体,可忽略,此时无论CT值多大均不影响数据
右上图:NTC熔解曲线与目的标本的峰图重合→NTC有污染,此时NTC的CT比目的基因大3个或者5个循环才可以用。

NRT的CT比目的基因大3个或者5个循环才可以用。
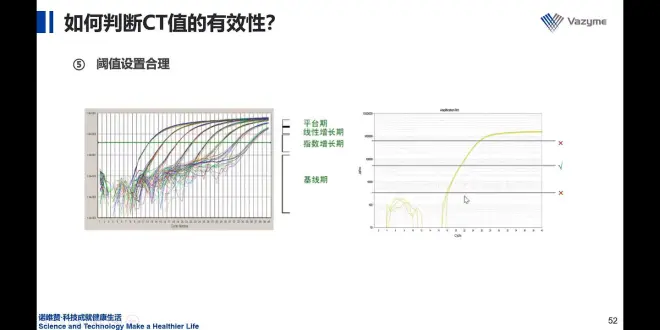
5.合理设置阈值,放在指数增长期
左图:阈值放在中间,接近于直线的区域

内参中的e相近于1,才可以计算。
e:扩增效率

二)数据处理
1.相对定量


1.绝对定量

标准品很重要,
CDNA不能做标准品原因:浓度不准确
四、F&Q
一)图一


二)图二
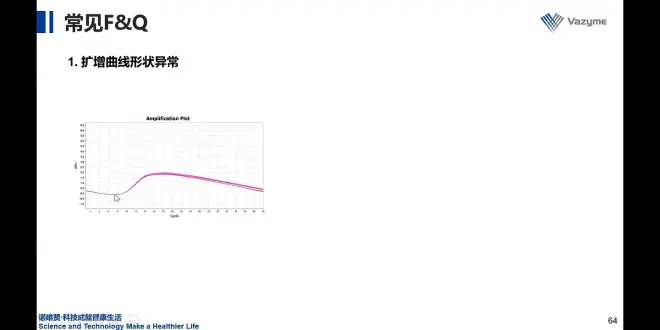
基线设置错误/模板浓度过高,基线期一般机器默认,但模板投入过多时默认的基线期不再适合,可手动调整

15调整为10

三)图三

上图:仪器程序设置问题
下图:定期维护矫正


左上:主峰前出现杂峰→引物特异性不好/引物二聚体/引物浓度过高/模板浓度过低
右上:主峰后出现双杂峰→交叉污染/引物特异性差——重新设计引物/对操作环境清洁
左下:Tm<80℃→扩增片段过短/只有引物二聚体无目的产物——重新设计引物/尝试提高模板浓度退火温度/降低引物浓度/pcr产物电泳检测有无条带确定模板投入是否正确
右下:熔解曲线峰高低

