
USP22通过去泛素化输入蛋白KPNA2来促进IRF3核易位和抗病毒反应
写在前面
今天推荐的是由武汉大学中南医院生命科学学院病毒学系在2020年1月13日发表于Journal of Experimental Medicine(2020IF:14.307,JCRQ1)的一篇文章,通讯作者是Bo Zhong教授,研究表明USP22通过去泛素化输入蛋白KPNA2来促进IRF3核易位和抗病毒反应。
研究背景
去泛素化酶 (DUB) 从靶蛋白中去除多聚或单泛素缀合物,从而调节其稳定性或活性。泛素特异性蛋白酶 (USP) 22属于DUB的USP亚家族,与小鼠胎盘发育和人类癌症有关。
摘要部分
在这里,作者发现细胞质USP22通过在病毒感染后去泛素化和稳定KPNA2来促进IRF3的核易位。病毒感染以依赖KPNA2的方式诱导细胞质中的USP22-IRF3结合,并且敲低或敲除USP22或KPNA2会损害病毒感染后IRF3核易位和下游基因的表达。一致地,与相应的对照同窝仔鼠相比,Cre-ER Usp22fl/fl或Lyz2-Cre Usp22fl/fl小鼠在病毒感染后产生的I型干扰素水平降低,并且对致命病毒感染的易感性增加。从机制上讲,USP22在病毒感染后使KPNA2去泛素化并稳定化,以促进IRF3的有效核易位。将KPNA2重组为USP22敲除细胞,恢复了病毒引发的IRF3核易位和细胞抗病毒反应。这些发现定义了细胞质USP22以前未知的功能,并在USP22和IRF3核易位之间建立了联系,从而扩展了传染病的潜在治疗策略。
研究内容
1.USP22与病毒感染后的IRF3相关
IRF3是一种转录因子,可在病毒感染后介导大量基因的表达。作者通过免疫共沉淀和免疫印迹分析来筛选与IRF3相互作用的DUB。最终将USP22鉴定为IRF3相互作用的DUB。在平行的报告基因筛选试验中,USP22激活仙台病毒(SeV)诱导的干扰素刺激反应元件(ISRE)启动子的激活。USP22位于细胞质和细胞核中,并且USP22的敲低会损害病毒触发的下游基因表达。USP22在IRF3的调控方面比其他DUB有更高的可能性。因此作者选择了USP22进行进一步研究。作者接下来检查了IRF3和USP22之间的内源性关联,免疫印迹分析显示,在SeV或HSV-1感染后,USP22与小鼠BMDC和MEF中的IRF3相互作用。据报道,USP22位于细胞质和核质中。作者观察到,在BMDC中水泡性口炎病毒(VSV)或HSV-1感染后,USP22与细胞质中的IRF3和磷酸化IRF3(pIRF3)相互作用,但不在细胞核中相互作用。N端的四个碱性氨基酸残基(163-KRRK-166)是USP22核定位所必需的NLS。有趣的是,作者发现USP22 (RR164/165AA)仍然与IRF3相关。域映射分析的结果表明USP22的C端泛素肽酶域(aa169-525)负责它们的关联。

研究结论:病毒感染后USP22与细胞质中的IRF3相互作用。
2.USP22的敲低抑制人细胞系中的RF3核聚集
为了确定USP22是否是IRF3的生理调节剂,作者设计了靶向USP22的siRNA。定量逆转录qRT-PCR分析的结果表明,USP22的敲低抑制了SeV或HSV-1诱导的THP-1细胞中IFNB、ISG56和CCL5的表达以及SeV诱导的IFNB、ISG56和HeLa细胞中的CCL5。先前的研究表明,USP22 与 ENY2 和 ATXN7L3 相互作用并形成转录共激活因子 SAGA 复合物,以调节 H2B 单泛素化水平。然而,敲除ENY2或ATXN7L3对SeV诱导的IFNB、ISG56和CXCL10在HeLa细胞中的表达没有明显影响,表明USP22以独立于SAGA复合物形成的方式调节人类细胞系中病毒触发的下游基因表达。IRF3的磷酸化和二聚化是病毒感染后IRF3激活的两个标志。然而,SeV或HSV-1诱导的IRF3磷酸化并未因THP-1细胞中USP22的敲低而受损。此外,USP22的过表达和敲低均不影响病毒诱导的IRF3在THP-1或HeLa细胞中的二聚化。另一方面,作者发现在SeV或HSV-1感染后,USP22的敲低显着削弱了IRF3在THP-1细胞中的核积累。这些数据共同表明USP22参与病毒感染后IRF3的核积累。

研究结论:USP22参与病毒感染后IRF3的核积累。
3.敲除小鼠原代细胞中的USP22会削弱病毒触发的信号传导
USP22与小鼠的早期胚胎发育和神经退行性疾病有关,人类癌症和小鼠USP22的胚系缺失会导致早期胚胎缺陷。为了进一步研究USP22在体内抗病毒信号传导中的作用,作者成功构建USP22敲除小鼠(Cre-ER Usp22fl/fl),该小鼠具有正常的免疫细胞稳态。

作者接下来检查了各种4-羟基三苯氧胺(4-OHT)处理的Cre-ER Usp22fl/+和Cre-ER Usp22fl/fl细胞中病毒触发的下游基因诱导。正如预期的那样,与Cre-ER Usp22fl/+对应物相比,4-OHT处理的Cre-ER Usp22fl/flBMDC和骨髓源性巨噬细胞(BMDM)中Usp22的表达减少。qRT-PCR分析的结果表明,敲除USP22会显着削弱SeV-、VSV-、HSV-1-或转染的poly(I:C)-、细胞质DNA-和LPS诱导的Ifnb、Isg15和Ccl5在BMDC和BMDM中的表达。此外,BMDC中USP22的敲除显着削弱了细胞培养物上清液中VSV-、HSV-1-或配体诱导的IFN-β和CCL5的产生。与在人类细胞系中观察到的结果一致,SeV或HSV-1诱导的IRF3磷酸化或二聚化不受BMDC或MEF中USP22敲除的影响。相反,敲除BMDC或MEF中的USP22显着抑制了IRF3在细胞核中的积累,表明USP22促进病毒触发的IRF3核易位。与这一观点一致,BMDC或MEF中USP22的敲除显着促进了野生型或GFP标记的VSV或HSV-1复制,如噬菌斑测定和流式细胞术分析所示。

作者还通过杂交Usp22fl/+小鼠和Lyz2-Cremice获得了Lyz2-Cre Usp22fl/+和Lyz2-Cre Usp22fl/fl小鼠。与Cre-ER Usp22fl/fl细胞中的发现相似,Lyz2-Cre介导的USP22缺失显着削弱了SeV或HSV-1诱导的Ifnb和Isg15在BMDC中的表达。此外,与Lyz2-Cre Usp22fl/+对应物相比,SeV-或HSV-1诱导的IRF3核易位在Lyz2-Cre Usp22fl/fl BMDCs或BMDMs中受到抑制。通过流式细胞术和荧光显微镜成像,与Lyz2-Cre Usp22fl/+BMDCs相比, VSV-GFP或H129-G4的复制在Lyz2-Cre Usp22fl/fl BMDCs中显着增强。

研究结论:USP22对于病毒触发的IRF3核易位和随后的细胞抗病毒反应至关重要。
4.USP22缺陷小鼠对病毒感染的易感性增加
为了表征USP22在体内RNA病毒感染中的作用,作者连续5天将他莫昔芬(80mg/kg)腹腔注射到Cre-ER Usp22fl/+和Cre-ER Usp22fl/fl小鼠中,7天后,小鼠通过尾静脉或腹腔注射感染VSV或HSV-1。Cre-ER Usp22fl/fl小鼠比对照同窝仔鼠更容易受到致死性VSV感染。此外,在VSV感染后12小时,对照小鼠相比,Cre-ER Usp22fl/fl小鼠血清中IFN-β和CCL5的浓度显着降低。在VSV感染后24小时或4天,与Cre-ER Usp22fl/+小鼠相比,来自Cre-ER Usp22fl/fl小鼠的肺或脑中Ifnb、Isg15和Ccl5的表达严重受损。一致地,在腹腔注射VSV后4天,Cre-ER Usp22fl/fl小鼠的脑或肾脏中的VSV滴度显着高于Cre-ER Usp22fl/+小鼠。同样,作者观察到敲除USP22导致对致死性HSV-1感染的易感性增加,并降低HSV-1感染后血清中IFN-β和CCL5的产生。此外,与Cre-ER Usp22fl/+小鼠相比,在HSV-1感染后24小时或4天,来自Cre-ER Usp22fl/fl小鼠的肺或脑中Ifnb、Isg15和Ccl5的表达分别严重受损。通过敲除USP22,大脑或脾脏中的HSV-1滴度显着增加。类似地,Lyz2-Cre对USP22的缺失导致对致死性VSV或HSV-1感染的易感性增加并减少VSV或HSV-1感染后血清中IFN-β和CCL5的产生。

研究结论:USP22正调节病毒诱导的下游基因表达,并且对于宿主体内针对病毒的防御至关重要。
5.USP22的酶活性是抗病毒信号传导所必需的
作者接下来检查了USP22介导的抗病毒信号传导激活是否需要核定位或去泛素化活性。将空载体、USP22、NLS突变体USP22 (RR164/165AA)或酶失活突变体USP22 (C185S)重组到Cre-ER Usp22fl/fl细胞中,然后进行4-OHT处理和SeV、VSV或HSV-1感染。qRT-PCR和ELISA分析的结果表明,病毒诱导的Ifnb、Isg15、Ccl5或Isg56表达以及IFN-β和CCL5的产生在用USP22或USP22(RR/AA)重组的Cre-ER Usp22fl/flMEF中得到显着恢复但在用USP22(C185S)重组时则没有这种效果,表明酶活性而非USP22的核定位是病毒触发信号传导所必需的。此外,通过将USP22或USP22(RR/AA)而非USP22(C185S)重组为Cre-ER Usp22fl/flMEF,SeV或HSV-1诱导的IRF3核易位增加。一致地,在用USP22重建的Cre-ER Usp22fl/flMEF中,VSV和HSV-1的复制也显著减少。
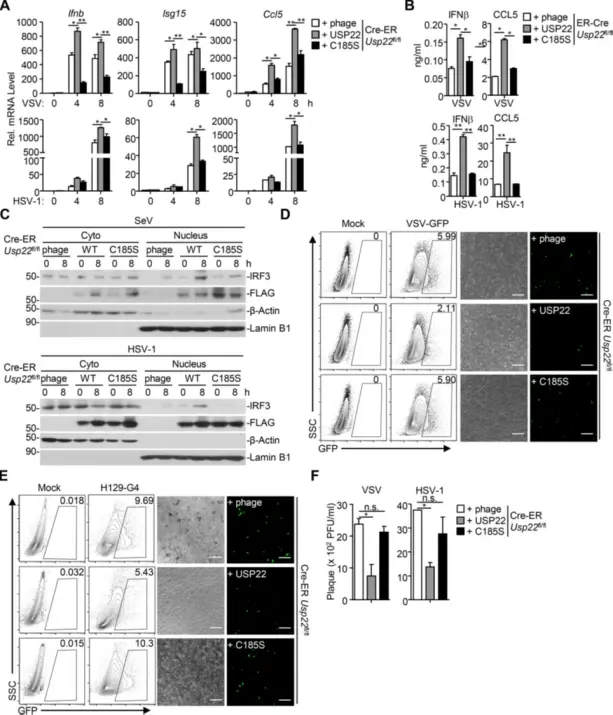
研究结论:USP22介导的病毒触发信号需要其去泛素化酶活性。
6.USP22在IRF3水平起作用,但不针对IRF3进行去泛素化
因为USP22介导的病毒触发信号需要其酶活性并且USP22与IRF3相互作用,作者认为USP22可能催化IRF3的去泛素化并调节IRF3的核易位。然而,作者发现SeV或HSV-1诱导的IRF3泛素化在4-OHT处理的Cre-ER Usp22fl/+和Cre-ER Usp22fl/flMEF之间具有明显差异。此外,在VSV感染后,敲除USP22对IRF3的稳定性影响很小。USP22的过表达或敲低分别增强或抑制了IRF3(5D)介导的ISRE报告基因的激活,而USP22(RR/AA)而不是USP22(C185S)的过表达促进了IRF3或IRF3(5D)介导的ISRE激活,表明细胞质中USP22酶活性在促进病毒触发的信号传导方面起主要作用。先前的研究表明,磷酸酶和张力蛋白同源物(PTEN)介导的IRF3在Ser97处的去磷酸化促进了其核易位。作者发现USP22的过表达增强了IRF3(S97A)介导的ISRE激活。

研究结论:USP22以依赖于其酶活性的方式促进病毒诱导的IRF3在细胞质中的核易位。
7.USP22去泛素化并稳定KPNA2
内源性免疫沉淀试验的结果表明KPNA2与USP22组成型相互作用,但与THP-1细胞中的IRF3或pIRF3相互作用,其方式取决于病毒感染。KPNA2的敲低削弱了MEF中病毒诱导的USP22-IRF3关联,表明USP22在病毒感染后通过KPNA2与IRF3相互作用。作者接下来检查了在有无病毒感染的情况下,USP22充足或缺乏的MEF中KPNA2的泛素化。结果表明,病毒诱导的KPNA2泛素化通过敲除USP22得到显着增强,并且将USP22而非USP22(C185S)重构到USP22缺陷型MEF时,减少了KPNA2的泛素化。此外,USP22 或 USP22(RR164/165AA) 而不是 USP22(C185S) 在体外使 KPNA2 去泛素化,表明USP22直接催化KPNA2的去泛素化。正如预期的那样,在VSV感染后,USP22缺陷的MEF中KPNA2的半衰期比USP22充足 的MEF短,这可以通过重构USP22而不是USP22(C185S) 逆转。在USP22敲除的MEFs中,KPNA2的加速降解被Bafilomycin A1或敲除ATG7完全挽救,但不被MG132挽救,表明USP22在病毒感染后去泛素化并保护KPNA2免受自噬依赖性降解。NDP52和p62是重要的选择性自噬受体,能识别并将多聚物修饰的目标运送到自噬体。有趣的是,敲除NDP52而不是p62完全抑制了USP22缺陷的MEFs中病毒引发的KPNA2的降解。

研究结论:USP22介导的KPNA2的去泛素化通过NDP52介导的选择性自噬途径抑制KPNA2的降解。
8.KPNA2介导病毒触发的IRF3核易位
作者接下来检查了KPNA2对病毒触发的信号传导和IRF3核易位的影响,发现KPNA2的敲低显着抑制了VSV或HSV-1诱导的Ifnb和Isg15在初级MEF或THP-1细胞中的表达。正如预期的那样,KPNA2的敲低大大削弱了SeV或HSV-1感染后MEF中IRF3的核转位,并促进了THP-1细胞中VSV或HSV-1的复制。这些数据表明KPNA2对于病毒诱导的IRF3核易位和细胞抗病毒反应至关重要。

正如预期的那样,在SeV或HSV-1感染后,将KPNA2重组到USP22敲除的MEFs中可恢复Ifnb和Ccl5的表达以及IRF3的核易位。一致的是,在用KPNA2重构的USP22敲除细胞中,VSV和HSV-1的复制被明显抑制,而在用空载体重组的细胞中则没有。

研究结论:USP22通过去泛素化和稳定KPNA2来促进病毒诱导的IRF3的核易位。
结论与讨论
在这项研究中,作者发现USP22在病毒触发的IRF3核易位和细胞抗病毒反应中的重要作用。USP22在病毒感染后以KPNA2依赖性方式与IRF3相互作用。USP22去泛素化并稳定KPNA2,从而促进病毒触发的IRF3核易位和下游基因的后续表达。病毒感染后,敲除USP22会损害lRF3的核积累,并增强病毒在体外和体内的复制,这可以通过KPNA2的重构来恢复。总的来说,这些发现定义了细胞质USP22以前未知的功能,并在USP22和IRF3核易位之间建立了机制联系,从而扩展了传染病的潜在治疗策略。
Thank you!
原文链接:https://doi.org/10.1084/jem.20191174
