
Autodock vina results analysis 对接结果分析
Autodock vina results Analysis 对接结果分析
菜鸟博士Caesar
对接结果分析:
results Analysis
1.载入对接结果
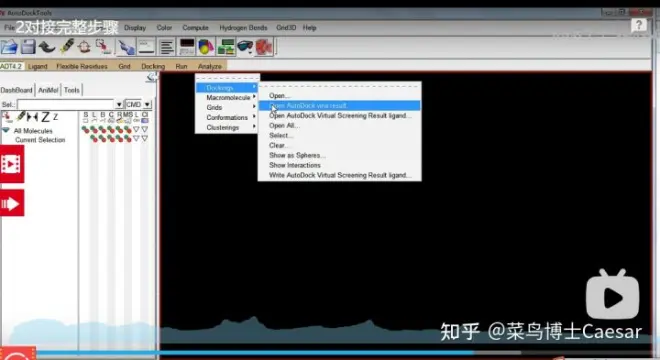
Analyze-Docking-Open AutDock vina result...载入对接结果(result.qbtqt),为方便观察,在弹出的对话框中选择载入模型的方式为“Single molecule with multiple conformations”,点击“确认”,可以看到窗口中能量最低的配体构象,通过键盘向右键,可以依次观察不同的对接结果(different conforms and energy);
2.载入受体
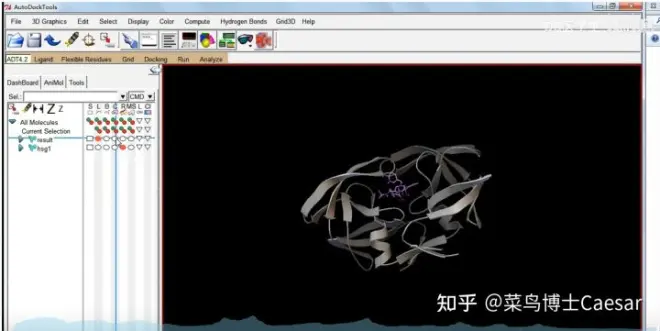
Analyze-Macromolecule-Open...载入受体
3.分析相互作用
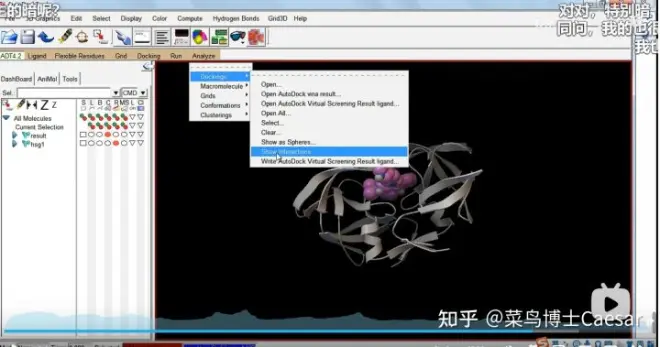
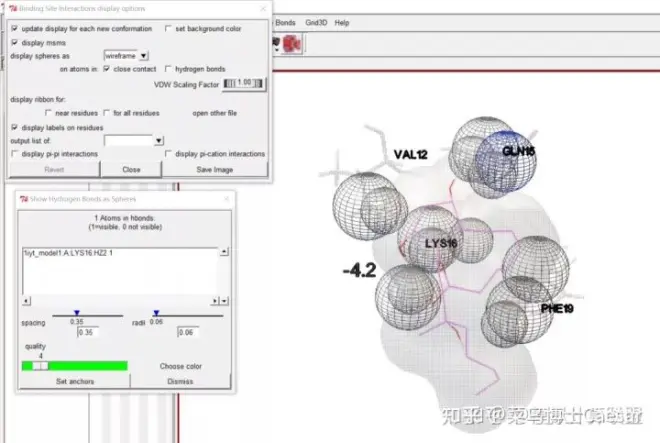
Analyze-Docking-Show Interactions,可以看出受体的Lys16与配体形成了1个氢键,分别勾选“display pi-pi interactions”和“display pi-cation interactions”,可以显示是否形成pi-pi相互作用和pi-C相互作用;
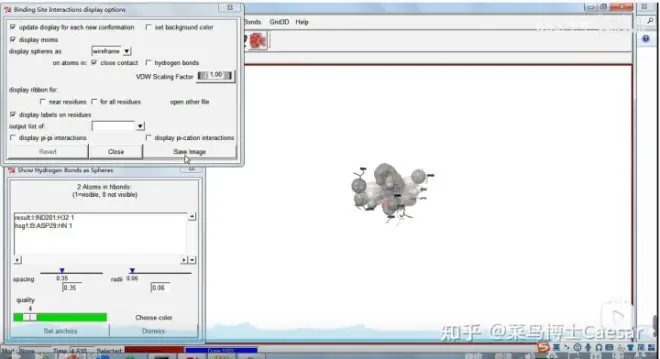
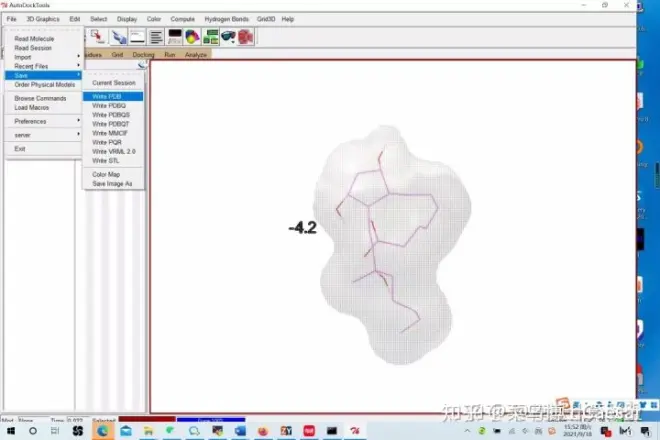
4.保存配体
右键删除受体蛋白,File-save Molecule-Write PDB,将符合要求的配体构象保存为PDB格式,用于Pymol作图;
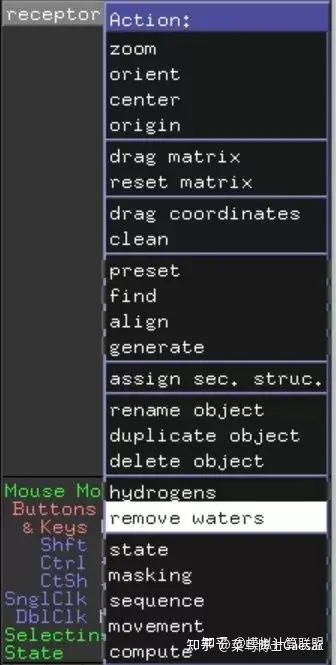
5.合并受体和配体分子
打开Pymol,File-Open依次载入受体、配体分子,点击Receptor-Action-remove waters去除受体中的水分子,
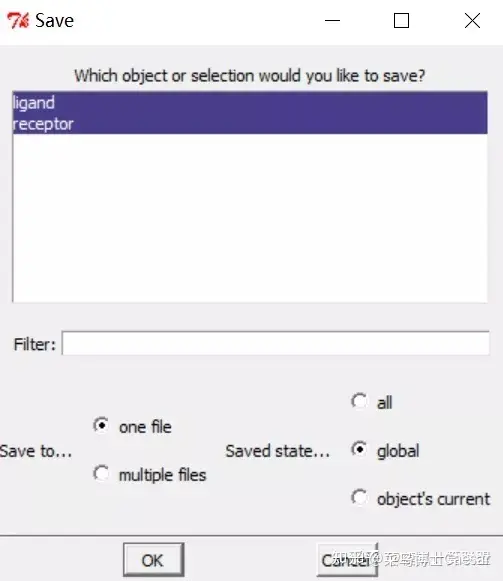
File-Save Molecule…,同时选择受体和配体,保存为complex.pdb;
6.显示氢键
File-Open载入刚刚的复合物complex.pdb,点击Ligand-Action-Find-polar contacts-to other atom in object,
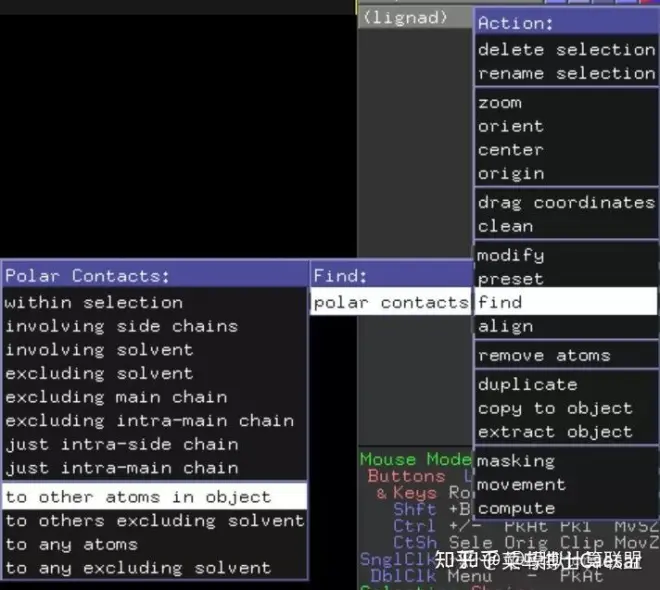
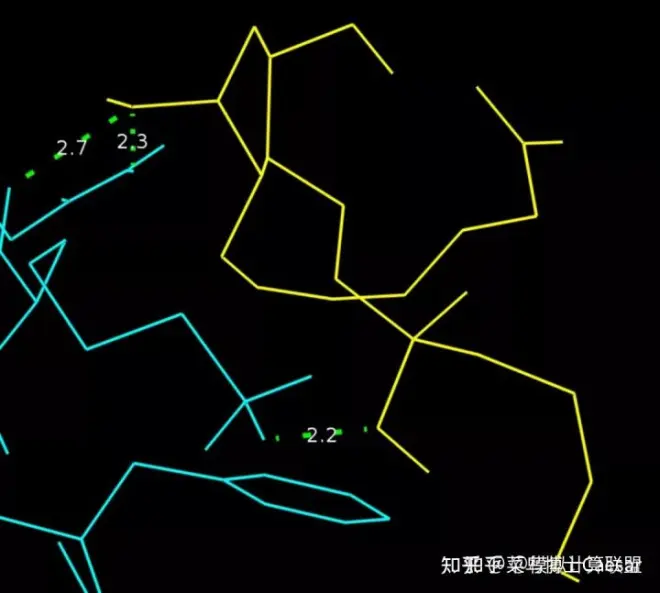
显示出如图的氢键作用,通过Show-labels,可显示出如图所示的氢键键长;
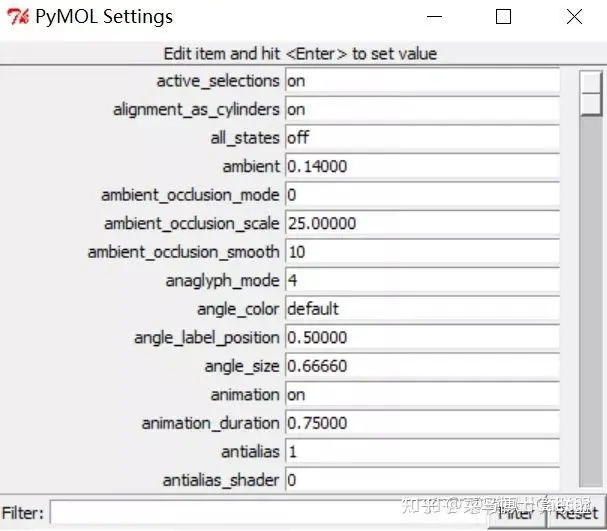
7.编辑氢键参数
在Externa GUI窗口的菜单栏中选择Setting-Edit All,可编辑各项参数值:
h_bond参数项,可更改氢键判断条件;
dash参数项,更改虚线的粗细、长度、颜色和间隔;
问题:show interaction可能报错memory error,大家可以一起讨论下
补充:计算RMSD pymol代码:align 2ORh_7O9Wout_model1,2ORh_7O9Wout1
(部分图片内容源于网络)
